
Contact

 +49-9131-85-27775
+49-9131-85-27775
 +49-9131-85-27270
+49-9131-85-27270
Secretary
| Monday | 8:00 - 12:15 |
| Tuesday | 8:00 - 16:45 |
| Wednesday | 8:00 - 16:45 |
| Thursday | 8:00 - 16:45 |
| Friday | 8:00 - 12:15 |
Address
Lehrstuhl für Informatik 5 (Mustererkennung)
Martensstr. 3
91058 Erlangen
Germany
Powered by

Pattern Recognition Lab
Department of Computer Science 5
Researchers and students at Pattern Recognition Lab (LME) work on the development and implementation of algorithms to classify and analyze patterns like images or speech. The research is mostly interdisciplinary and is focussed on medical- and health engineering. The LME has close national and international collaborations with other universities, research institutes and industrial partners.
A summary of the projects at the Pattern Recognition Lab is available for download as a comprehensive brochure (PDF).
-
Research Areas
-
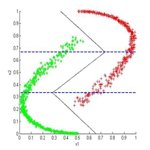
Pattern Recognition deals with the automatic classification and analysis of sensor input data. In this area we have work groups in
 Speech Recognition and Understanding, Computer Vision, Multiple Criteria Optimization, Image Analysis, Image Segmentation, and Image Fusion.
Speech Recognition and Understanding, Computer Vision, Multiple Criteria Optimization, Image Analysis, Image Segmentation, and Image Fusion.Medical Image Processing
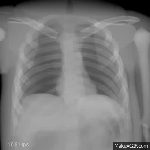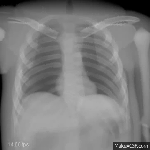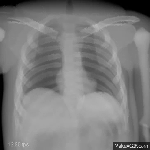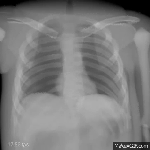
The research area medical image processing investigates formation and analysis of images in medicine. Currently the groups Analytic Reconstruction and Consistency,
Computed Tomography: Algebraic Reconstruction and Motion, Magnetic Resonance Imaging, Phase-Contrast Imaging, and Ophthalmic Imaging are working on such topics.
With the sheer amount of "big data", new opportunities are available also for pattern recognition and machine learning. Examples are
deep learning and  precision learning. In this area, we have the groups of Learning Approaches for Medical Big Data Analysis, Population Modelling, and Enterprise Computing.
precision learning. In this area, we have the groups of Learning Approaches for Medical Big Data Analysis, Population Modelling, and Enterprise Computing.
Available ThesesExtension of an audio-recordings database with features for similarity search (Master Arbeit) Betreuer: Maier, Andreas; Meyer-Wegener, Klaus
Recent PublicationsChoi, Jang-hwan; McWalter, Emily; Datta, Sanjit; Müller, Kerstin; Maier, Andreas; Gold, Garry; Levenston, Marc; Fahrig, Rebecca: In Vivo 3D Measurement of Time-dependent Human Knee Joint Compression and Cartilage Strain During Static Weight-Bearing In: ORS 2016 Annual Meeting
-
-
CooperationsLME-News
PhD student receives Luise Prell-Preis 2019
Franziska Schirrmacher, a former PhD student at the Pattern Recognition Lab, receives this year's Luise Prell-Preis for her excellent Master's thesis, from which publications evolved. Parts of the thesis are published in a MICCAI...[more]
Invited Talk by Prof. Michael King
On next Monday, Prof. Michael King from the University of Massachusetts Medical School will give a talk in our lab. Title: "Respiratory Motion Correction Methods for Cardiac SPECT and DL Denoising”Time: 15:00 on...[more]
Second Best Work at MS-CMRseg Challenge (STACOM 2019)

Sulaiman Vesal, PhD student at the Pattern Recognition Lab, was awarded the second best work in the STACOM Multi-sequence Cardiac MR Segmentation Challenge at MICCAI 2019. The award was given for the paper: "Automated...[more]
Five students excel in Deep Learning Class

Congratulations to Maniraman Periyasamy, Meike Biendl and Alexander Richter, Jonas Utz, and Henrik Willer for their outstanding achievements in the Deep Learning Challenge 2019! A great success. Please...[more]
New DFG Project on Deep Learning in Medical Imaging funded!

In a cooperation with Marc Kachelriess (DKFZ), Michael Lell (Klinikum Nürnberg Nord) and our lab, we successfully applied for another DFG Project. The aim of the project is to estimate patient-individualised dose for a CT scan...[more]
Best paper award at the Iberoamerican conference on pattern recognition (CIARP 2019)

Camilo Vasquez, a PhD student of our lab was warded with the best paper award at the Iberoamerican conference on pattern recognition (CIARP 2019) that was held in Havana (Cuba) from 28.10.2019 to 31.10.2019. The award was given...[more]
-

























Tired of doing research? Need a break? Need some good bavarian coffee? Check out the Bavarian Roasting Company and become a fan on Facebook.